
Feeling sick is difficult enough without going to a doctor’s appointment and learning that the diagnosis might possibly be the rare disease atypical hemolytic uremic syndrome, also known as atypical HUS (aHUS). Rushing home to seek out medical content on the internet with ‘Dr. Google’, it’s likely that information is difficult to find and hard to understand. Many patients face a more complex situation, that of hearing their doctor mention varied opinions as the medical team isn’t sure if the diagnosis is actually aHUS or rather a different medical condition that presents with similar symptoms. It’s a perfect storm for rare disease patients: vague symptoms that quickly become urgent, medical symptoms that are common to many different diagnoses, a syndrome few doctors have experience treating, and a very rare and complex disease that can affect multiple organs and body systems. Food borne illness sometimes receives media attention when food or water sources are contaminated by salmonella or E. Coli, causing an outbreak of regular HUS which shares similar symptoms to aHUS (Click Here FMI on food borne illness). aHUS families want to know disease basics, wish to gain a better understanding of conversations with their doctors, and hope to learn how their medical case history fits within such a complex disease diagnosis.
aHUS Basics
Atypical hemolytic uremic syndrome is a collection of symptoms, hence the term ‘syndrome’ in its name. A main feature of aHUS is the destruction of red blood cells, which explains use of the word ‘hemolytic’ since ‘hemo’ means blood and ‘lytic’ means disintegration of cells. Inclusion of the word ‘uremic’ means that rather than being filtered and removed, toxic waste products build up in the patient’s bloodstream, a sign of poor kidney function (renal disease) since waste products are normally excreted in urine. So why is ‘atypical’ part of the name of this very rare disease, estimated to affect 1 or 2 people out of a million? The more common form or ‘typical HUS’ usually happens when certain E. coli strains cause a gastrointestinal (GI) infection. Many forms of E. coli are normally present in the gut of humans and warm-blooded animals, but certain strains of E. coli are not and it’s these bacterial strains that release Shiga toxins that can cause diarrhea, intestinal bleeding and damage to the bowel walls and kidneys. Also known as D+HUS (or HUS associated with diarrhea), typical HUS may result from eating undercooked or poorly processed food products, or from exposure to water contaminated with certain forms of E. coli bacteria. Typical or D+HUS accounts for 95% of all cases, with the remaining 5% of HUS cases termed ‘atypical HUS’ to indicate a case not caused by bacteria. (Note: While aHUS is sometimes called D-HUS to note that lack of diarrhea as a symptom, a confusing factor in diagnosis is that approximately 25% of typical HUS or D+HUS patients actually present without any diarrhea symptoms.)
Patients with either aHUS or typical HUS would have these similar symptoms: blood and blood vessels are affected, red blood cell counts are low (anemia), platelet counts drop (used in clotting, thrombocytopenia is too few platelets) and tiny clots which may form anywhere in the body but often affect the kidneys and brain. Development of tiny clots (thrombi) can happen in different parts of the body to cause sudden or life-threatening conditions such as seizures, vision loss, stroke, cardiac issues, kidney failure and other serious medical situations. Typical HUS episodes are usually one-time infections, associated with food outbreaks or animal sources that have contaminated food or water supplies. Sometimes we just can’t figure out why the disease has occurred, and “idiopathic” HUS refers to illness that isn’t connected to any particular cause. Genetic aHUS (also known as hereditary HUS, familial aHUS, or complement mutation-associated HUS) may occur at any age and accounts for an estimated 60% of all aHUS cases. Triggers for acquired aHUS may include bacterial and viral infections. Some drugs may trigger aHUS such as certain chemotherapeutics, antiplatelet drugs, immunotherapeutics, and include common medications such as oral contraceptives and anti-inflammatory drugs. Pregnancy-associated aHUS may develop during at various stages including post-delivery, with transplantation and underlying medical conditions such as autoimmune diseases also known to trigger aHUS activity. In the case of atypical HUS versus other forms of HUS or other diseases with similar symptoms, are there differences in diagnosis, treatment, and management? In a word, yes.
What’s the difference between aHUS and Typical HUS (STEC-HUS)?
What basic information might be helpful to patients and their families to know as they listen to their medical team confer with them as to whether their diagnosis might be atypical or typical HUS? There are various tests (immunoassays) which require only a few hours to detect bacterial causes, and diagnosis of STEC-HUS (Shiga toxin-producing E. coli HUS) usually will involve testing a patient stool sample. Patients with typical HUS often respond well to supportive treatment such as infusions of red blood cells and dialysis to help their kidneys recover.
Supportive care for aHUS patients includes plasma infusion or plasma exchange (also called apheresis or plasmapheresis), with dialysis as needed for kidney injury or renal failure. While patient responses to plasma therapies may be mixed, it’s widely available around the globe and therefore often used as a first line medical treatment due to this broad availability. Eculizumab was the first approved therapeutic drug for treating patients with aHUS, followed in Oct 2019 by FDA approval of ravulizumab, for but eculizumab is not available in all nations and drug access may be restricted by governmental regulations by country or healthcare policy.
Patients may be referred by their primary care doctor to a physician with special medical training in areas such as: nephrology (kidney or renal diseases), hematology (blood disorders), immunology (immune issues, to include the complement system), or other specialty area (oncology or hem-onc for example). Since aHUS can be an inherited condition passed down in the family (genetic HUS or familial aHUS), sometimes geographic locations can have several aHUS cases grouped together. Otherwise, aHUS is so rare that most doctors never diagnose or treat a single case of atypical HUS in their medical careers. Exact numbers of aHUS patients are difficult to determine, but this mathematical approach can help determine an estimated worldwide number of aHUS cases. Currently the 2017 world population clock indicates about 7.5 billion people on earth, and aHUS literature refers to an estimated 1 to 2 atypical HUS patients per million population. Given that 7.5 billion people is equivalent to 7500 million, statistically there could be an estimated 7,500 to 15,000 people around the world with atypical HUS. Due to family inheritance of genetic factors among some aHUS patients, logic leads to the possibility of some so-called geographic ‘aHUS hotspots’ or regions with a small concentration of atypical HUS patients likely treated by the same specialty practice or physician in their region.
See this article’s section of research and resources for detailed information on aHUS diagnosis and treatment, and other related reference material.
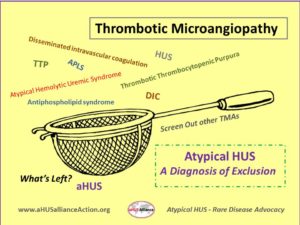
aHUS as a type of Thrombotic Microangiopathy
There are many ways that physicians and researchers compare diseases with similar characteristics, which may serve to confuse patients and caregivers trying to sort through medical information. Sometimes atypical HUS will be grouped with other medical conditions as a general category of ‘complement mediated kidney diseases’ and other times information about aHUS can be found under the heading of ‘thrombotic microangiopathies’. Around the world there are different spellings and abbreviations used for atypical HUS, making research more difficult for families to uncover. Here’s a very brief overview of terminology and common issues up for consideration as doctors sort through the diagnostic process for patients suspected to have aHUS.
In most cases aHUS is caused by uncontrolled activation of the complement system, part of the body’s immune system that we all are born with, and which usually acts in a controlled manner to defend against disease and inflammation to maintain good health. Atypical HUS affects patients of all ages, though its symptoms and their severity range widely, and aHUS frequently has a genetic component. A main characteristic of aHUS is the formation of tiny blood clots that systemically can occur throughout the body, known as ‘thrombotic microangiopathy’ (TMA). Raised lining of the small vessels and clots blocking them can shear red blood cells into pieces, which impacts their ability to carry oxygen from your lungs throughout the body. Red blood cells contain a protein called hemoglobin, and hemoglobin picks up oxygen in the lungs to release to tissues as blood flows through the body. Your bone marrow makes these red blood cells, but it’s difficult for the bone marrow to speed up production enough to replace lots of damaged red blood cells that have a shortened lifespan. ‘Hemolytic anemia’ occurs when a patient does not have enough healthy red blood cells (RBCs) created at a rate to keep pace with those being destroyed. (Note: Microangiopathic hemolytic anemia or MAHA is a more recent term used to designate any hemolytic anemia related to fragmentation of red blood cells.)
Platelets are an important component of your blood as these colorless cells help blood to clot, which stops cuts from bleeding when this clumping forms plugs. While helpful in case of injuries, random clot formation in blood vessels can block blood flowing as it should around the body and may cause life-threatening issues. If platelets break apart to form clots at an accelerated rate, patients can have low blood platelet counts, a condition called ‘thrombocytopenia’. The kidneys filter out liquid and waste from the blood, which is concentrated and removed from the body as urine. If clots occur in the kidneys they can clog up the tiny blood vessels (glomerulus) that are a key part of the structure of the kidney’s filtration units (nephrons). One product of muscle metabolism is creatinine, and this waste product is produced by the body at a fairly constant rate to be then removed from the blood stream by the kidneys. If kidney damage occurs from tiny clots, removal of waste products such as creatinine will be compromised (causing creatinine levels to rise) and these can build up to toxic levels unless eGFR (glomerular filtration rate) becomes assisted by machine through the process known as dialysis.
Patient care can be very complex when there is overlap, or when another disease state must be treated at the same time. Sometimes thrombotic microangiopathies (TMAs) are induced by or happen with an underlying medical condition such as cancer or autoimmune disorders (with aHUS secondary to SLE or lupus nephritis), or it may occur after stem-cell (HSCT) or organ transplantation (TA-TMA). ‘Secondary TMAs’ might trigger due to infections, be associated with pregnancy or delivery, or become activated by certain drugs, understandably with treatment needs focused on the cause or background disease. Genetic testing in the future may advance and expand to provide additional insight and information in this area. In some cases of atypical HUS, organs other than the kidneys might be most impacted. The term ‘extra renal involvement’ indicates disease activity that may affect organs other than kidneys, such as heart, lungs, GI tract, eyes, skin, or brain). This can make diagnosis of atypical HUS even more complicated, as physicians may encounter challenges when attempting to piece together facts and gain an accurate view of diagnosis and treatment.
Labs results that indicate low red blood cell or platelet counts, low amounts of hemoglobin, and reports of high creatinine levels may indicate atypical HUS activity – but they are also very common issues for patients with thousands of other medical conditions. Acute kidney damage may be caused by many things such as diet or diabetes, and many medical conditions can be a root cause of why kidneys might stop working to the extent that dialysis is needed to mechanically clean the blood (renal failure or end stage renal disease ESRD). Fatigue and mental confusion can occur even with a common cold or they might be features of a more serious health concerns; however an aHUS patients’ low hemoglobin or red blood cell counts might cause tiredness, and poor kidney function can make concentration and memory issues a lifestyle issue for those with atypical HUS. Simply put, lab test results may indicate general patient health trends and body processes but they cannot be used to diagnose atypical HUS.
Atypical HUS or Thrombotic Thrombocytopenic Purpura (TTP)?
For both aHUS and TTP, a key characteristic is the tiny clots (thrombotic microangiopathy or TMA) that can form throughout the body, in addition to other common features such as red blood cell destruction, low platelet counts and kidney injury. Patients with TTP additionally experience fever and neurological symptoms. In patients with TTP, the body doesn’t make enough of an enzyme called ADAMTS13, which cuts large proteins called von Willebrand factor into smaller pieces. Since von Willebrand factor helps platelets clot together and stick to blood vessel walls, having adequate amounts of ADAMTS13 prevents those big proteins from forming unnecessary blood clots. Diagnosis of TTP involves an ADAMTS13 test (assay) to determine its activity percent, with low results noted in about 80% of patients with thrombotic thrombocytopenic purpura (TTP). Sometimes aHUS patients have a partial ADAMTS13 deficiency, further emphasizing that atypical HUS is often a ‘diagnosis of exclusion’ as doctors rule out other diseases until the only diagnosis left is aHUS.
As a group of international aHUS patient organizations, the aHUS Alliance has gathered research and created resources to provide this article as a mere starting point – a basic overview of information related to atypical HUS, diagnosis and treatment of aHUS, and reference materials that may help distinguish aHUS from other thrombotic microangiopathies. Atypical HUS presents very differently in each patient, so while patient education and family support is very important, know that only medical professionals can work with patients and families to determine the appropriate diagnosis and a personalized treatment plan. We encourage readers to explore, question, and learn more about atypical HUS – and invite you to connect globally and collaborate with the aHUS Alliance.
L Burke April 2017
Last Update: July 2020
New information about aHUS diagnosis, thrombotic microangiopathy, and other topics is available on our website, in our Research & Publications section with related assets on our Resources page.
Main Source of Information for this Article
Genetic Atypical Hemolytic-Uremic Syndrome (NIH GeneReviews)
Detailed explanation of aHUS, with links to click for definitions and research.
GENERAL Info & Facts about aHUS
aHUS Info Centre
aHUS Resource Page
aHUS Facts- a Brief Look (Sept 2019) One Double-Sided Page, to Print & Share
Know aHUS: Know Us Living with aHUS (available in ES, EN, FR)
aHUS Video – Clinical Presentations
Atypical HUS Clinical Channel Video presentations by aHUS researchers, clinicians, and organizations on topics such as diagnosis, the role of complement, genetics, thrombotic microangiopathy, pregnancy, and more.
Thrombotic Microangiopathy Symposium: Through the Lens of aHUS 9 Videos featuring clinical presentations at a medical education event in Boston, each with a ‘Patient Voice’ segment to provide an authentic view of differential diagnosis, multidisciplinary team approach to care, treatment considerations, pregnancy & aHUS, and challenges for patients & physicians.
For Medical Teams treating a Patient Suspected to have aHUS
Atypical HUS: Critical Care (Diagnosis, Differentiating TMAs, Organ Involvement, Treatment)
Azoulay E et al. Expert Statements on the Standard of Care in Critically Ill Adult Patients With Atypical Hemolytic Uremic Syndrome. Chest Journal, 2017. Volume 152, Issue 2, Pages 424–434
Link to MORE on this topic: Research & Publications
aHUS Diagnosis & Treatment
Campistol JM et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia 2013;33(1):27-45
Cataland, S and Wu HM. How I treat: the clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome. ASH: Blood First Edition paper, March 5, 2014; DOI 10.1182/blood-2013- 11-516237
Gavriilaki E et al. Modified Ham test for atypical hemolytic uremic syndrome. Blood Jun 2015, 125 (23) 3637-3646; DOI: 10.1182/blood-2015-02-629683
Kaplan BS et al. Current treatment of atypical hemolytic uremic syndrome. Intractable Rare Dis Res IRDR 2014 May; 3(2): 34–45. doi: 10.5582/irdr.2014.01001
Laurence J et al. Atypical Hemolytic Uremic Syndrome (aHUS): Essential Aspects of an Accurate Diagnosis. Clinical Advances in Hematology & Oncology Volume 14, Issue 11, Supplement 11, November 2016
Magro C et al. The Role of the Skin Biopsy in the Diagnosis of Atypical Hemolytic Uremic Syndrome. Am J Dermatopathol. 2015 May; 37(5): 349–359 doi: 10.1097/DAD.0000000000000234
Nester CM and Thomas CP. Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated? doi:10.1182/asheducation-2012.1.617 ASH Education Book December 8, 2012vol. 2012 no. 1 617-625
Scully M and Goodship TH. How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol. 2014 Mar; 164(6): 759–766. doi: 10.1111/bjh.12718
Shin Yu-Min. Clinical evaluation of thrombotic microangiopathy: identification of patients with suspected atypical hemolytic uremic syndrome. Thromb J. 2016; 14(Suppl 1): 19. 2016 Oct 4. doi: 10.1186/s12959-016-0114-0
Link to MORE on this topic: Research & Publications
aHUS Guidelines – Consensus Documents
Azoulay E et al. Expert Statements on the Standard of Care in Critically Ill Adult Patients With Atypical Hemolytic Uremic Syndrome Chest 2017 Aug;152(2):424-434. doi: 10.1016/j.chest.2017.03.055.
Goodship THJ et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney International, Volume 91 , Issue 3 , 539 – 551. Dec 2016
Loirat C et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 16 Feb 2015 doi: 10.1007/s00467-015-3076-8
Link to MORE on this topic: Research & Publications
Typical HUS, STEC-HUS
Noris M et al. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nature Reviews Nephrology 8, 622-633 (November 2012) doi:10.1038/nrneph.2012.195
Sallée M et al. Thrombocytopenia is not mandatory to diagnose haemolytic and uremic syndrome. BMC Nephrology 201314:3 doi: 10.1186/1471-2369-14-3
Salvadori M and Bertoni E. Update on hemolytic uremic syndrome: Diagnostic and therapeutic recommendations. World J Nephrol 2013 Aug 6; 2(3): 56–76. doi: 10.5527/wjn.v2.i3.56
Wijnsma J et al. Unusual severe case of hemolytic uremic syndrome due to Shiga toxin 2d-producing E. coli O80:H2 Pediatric Nephrology, PuMed Journals: 28 March 2017
Wong C et al. The Risk of the Hemolytic–Uremic Syndrome after Antibiotic Treatment of Escherichia coli O157:H7 Infections. N Engl J Med 2000; 342:1930-1936June 29, 2000 DOI: 10.1056/NEJM200006293422601
ADAMTS-13
Feng S et al. Partial ADAMTS13 deficiency in atypical hemolytic uremic syndrome. Blood 2013 Aug 22; 122(8): 1487–1493. doi: 10.1182/blood-2013-03-49242
Phillips E.H et al. The role of ADAMTS‐13 activity and complement mutational analysis in differentiating acute thrombotic microangiopathies. J Thromb Haemost 2016 Jan; 14(1): 175–185. doi: 10.1111/jth.13189
TTP – Thrombotic Thrombocytopenic Purpura
Blombery P and Scully M. Management of thrombotic thrombocytopenic purpura: current perspectives J Blood Med 2014; 5: 15–23. doi: 10.2147/JBM.S46458
Cataland S and Wu H. Atypical hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: Clinically differentiating the thrombotic microangiopathies. Eur J Intern Med. 2013 Sep;24(6):486-91. doi: 10.1016/j.ejim.2013.05.007.
Scully M and Goodship T. How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol. 2014 Mar; 164(6): 759–766. doi: 10.1111/bjh.12718
Trachtman, H. HUS AND TTP Pediatr Clin North Am. 2013 Dec; 60(6): 1513–1526. doi: 10.1016/j.pcl.2013.08.007
Zheng XL. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med. 2015;66:211-25. doi: 10.1146/annurev-med-061813-013241.
Thrombotic Microangiopathies – TMAs
Appel G. Thrombotic microangiopathies: Similar presentations, different therapies. Cleveland Clinic Journal of Medicine. 2017 February; 84(2):114-116, 126-130
Al-Nouri Z et al. Drug-induced thrombotic microangiopathy: a systematic review of published reports Blood. 2015 Jan 22; 125(4): 616–618. doi: 10.1182/blood-2014-11-611335
Brocklebank V et al. Thrombotic Microangiopathy and the Kidney. Clin J Am Soc Nephrol. 2017 Oct 17. pii: CJN.00620117. doi: 10.2215/CJN.00620117
Chang, Anthony. Thrombotic microangiopathy and the kidney. Diagnostic Histopathology , Volume 23 , Issue 3 , 101 – 108
Eskazan A and Salihoglu A. Treatment and Outcome of Primary and Secondary Thrombotic Microangiopathies. Am J Nephrol. 2015; 41(6):427-8. doi: 10.1159/000437002. Epub 2015 Jul 14.
Feng S et al. Complement activation in thrombotic microangiopathies. Br J Haematol. 2013 Feb; 160(3): 404–406 doi: 10.1111/bjh.12112
George J and Nester C. Syndromes of Thrombotic Microangiopathy. N Engl J Med 2014;371:654-66. doi: 10.1056/NEJMra1312353
Gordon CE et al. Thrombotic Microangiopathy: A Multidisciplinary Team Approach. Am J Kidney Dis. 2017 Nov;70(5):715-721. doi: 10.1053/j.ajkd.2017.05.017.
Jokiranta TS et al Differential diagnosis of thrombotic microangiopathy in nephrologyBMC Nephrol. 2017; 18: 324. Published online 2017 Oct 28. doi: 10.1186/s12882-017-0727-y
Nguyen T and Han Y. Plasma exchange therapy for thrombotic microangiopathies. Organogenesis 2011 Jan-Mar; 7(1): 28–31. doi: 10.4161/org.7.1.14027
Ruggenenti P et al. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney International: Volume 60, Issue 3, September 2001, pgs 831–846
Link to MORE on this topic: Research & Publications
Link to more aHUS Alliance Articles on TMA

{kind=link}